It is important for you to know about following regulations, guidance and standards organizations as these regulatory authorizes govern and direct much work of the SAS programmer in the pharmaceutical industry.
International Conference on Harmonization (ICH)
Click to show/hide text
During the next 20 years, a lot of laws, regulations and guidelines for reporting and evaluating data on safety, quality and efficacy of new medicinal products emerged. And at the same time, the industry found that it was necessary to duplicate many time-consuming and expensive test procedures in order to market new products internationally due to the divergence in technical requirements from country to country.
The urgent need to rationalize and harmonize regulation was impelled by the concerns over rising costs and the need to minimize time required to launch new treatments available to patients in need. The success achieved by the European Community (now the European Union) in 1980s leaded to the birth of ICH at a meeting attended by representatives from Europe, US and Japan in April1990.
It aimed to define a common set of regulations so that a pharmaceutical regulatory application in one country can also be used in another. The comprehensive ICH guidelines are divided into four categories: Quality Guidelines, Safety Guidelines, Efficacy Guidelines and Multidisciplinary Guidelines. Of particular interest to SAS programmers are ICH E3 and ICH E9.
ICH E3 (Structure and Content of Clinical Study Reports) describes the format and content of a study report that will be accepted in all three ICH regions. ICH E9 (Statistical Principles for Clinical Trials) is more useful for Biostatistician as it describes essential considerations on the design and analysis of clinical trials. But it can give you an excellent overview of how a clinical trial should be conducted.
Clinical Data Interchange Standards Consortium (CDISC)
Click to show/hide text
SDTM defines data tabulation data sets to be sent to the FDA as part of a regulatory submission. It is usually the source data for creating ADaM datasets. CDISC ADaM are designed for creating statistical summaries and analysis. ODM is XML-based data model that allows for XML-based transmission of any data involved in the conduct of clinical trials. Define.xml is based on the ODM model and intended to be machine-readable. It is a replacement for define.pdf. The machine-readable property can allow FDA to work more easily.
Below figure shows you how to find all of the available standards in CDSIC website.
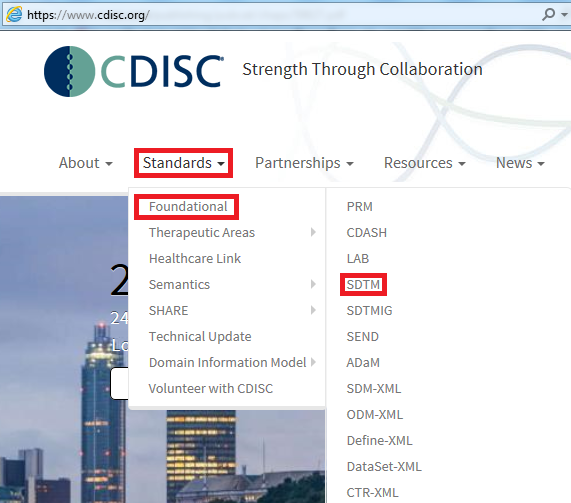
Food and Drug Administration (FDA) Regulation and Guidance
Click to show/hide text
Medicines & Healthcare products Regulatory Agency (MHRA)
Click to show/hide text